
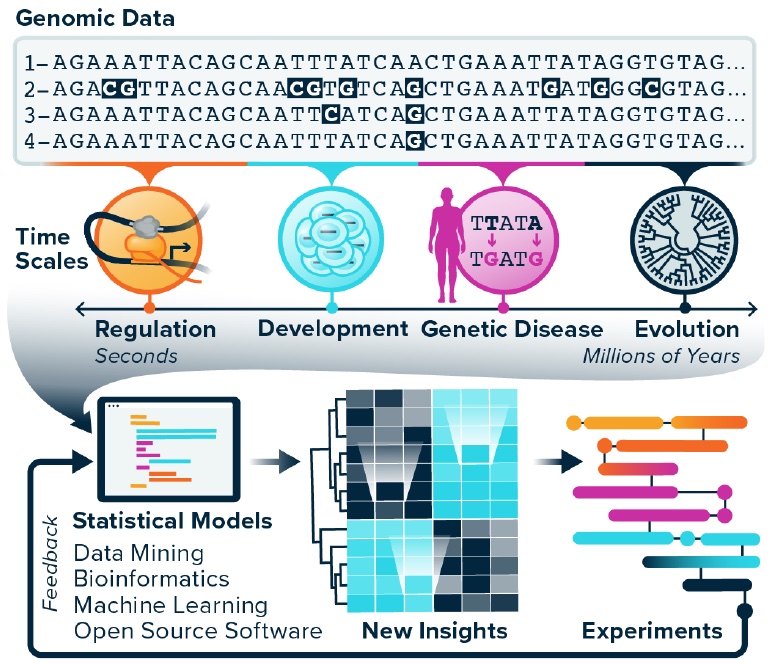
Our group studies how genomes work, evolve, and break in disease… We develop bioinformatics methods and models for comparative genomics. These tools help us to interrogate how genomes and their encoded functions change over time scales from developmental transitions in human cells to evolution across the tree of life. The methods we create are unified by statistical rigor, a phylogenetic perspective, and massive data integration. Collaboration, creating open source software, and promoting open science are priorities. Many of our projects involve machine learning, longitudinal modeling, and sequence analysis. Our NIH funded projects are listed on RePORTER.
Projects:
Biaswas Center for Transformative Computational Cancer Biology
Keck Center for Machine-Guided Functional Genomics
Regulatory genomics: The lab has several projects related to predicting and validating regulatory enhancers and investigating the role of fine-scale chromatin organization in gene regulation across evolution and disease.
- Methodology: Machine-learning, polymer simulations, motif models, clustering
- Ongoing work: Evolution of chromatin boundaries, functional characterization of enhancers and enhancer mutations
Human Accelerated Regions (HARs): We pioneered a statistical phylogenetic approach to identify the fastest evolving regions of the human genome (list) and showed that many of these sequences are developmental enhancers.
-
- Methodology: Continuous time Markov models, machine-learning
- Ongoing work: Massively parallel reporter assays and CRISPR screens of HARs, fast evolving DNA in other lineages and in cancer
- Lists of accelerated regions:
- 312 high-confidence HARs and 141 chimpanzee ARs identified by us using the Zoonomia Cactus alignments (Keough et al. Science, 2023)
- Merged list of 2649 HARs from us and other labs (Capra et al. PTRSB, 2013)
- Merged list of 721 HARs identified by us using MultiZ alignments (Pollard et al. Nature 2006; Pollard et al. PLoS Genetics 2006; Lindblad-Toh et al. Nature 2011)
- Human acceleration in mammal conserved regions (Lindblad-Toh et al. Nature 2011)
- Human acceleration in primate conserved regions (Lindblad-Toh et al. Nature 2011)
- Primate acceleration in mammal conserved regions (Lindblad-Toh et al. Nature 2011)
Metagenomics: We are designing methods to study the human microbiome and other microbial communities at the resolution of individual genes and genetic mutations.
- Methodology: Population genetics, phylogenetic regression, ecological statistics, bootstrap
- Ongoing work: Rapid metagenotyping, longitudinal microbiome dynamics, phylogenetically aware tests for association with host traits
Collaborations:
4D Nucleome – understanding the principals underlying dynamic 3D genome folding in development and disease using deep learning models
IGVF – decoding how genetic variation impacts genome functions
Zoonomia – discovering the genomic basis for shared and lineage specific traits in mammals
Chan Zuckerberg Biohub Microbiome Initiative – engineering the human microbiome to reveal its role in nutrition, immune function, and drug metabolism
PsychENCODE – Massively parallel characterization of psychiatric disease associated regulatory elements in defined cell types
B2B – Bench to Bassinet: The epigenetic landscape of heart development
BioFulcrum – team science projects to overcome disease
Gladstone Bioinformatics Core – provides expertise on experimental design and analysis of complex data sets, with a specialization on large-scale data sets acquired from various cutting-edge technologies.